
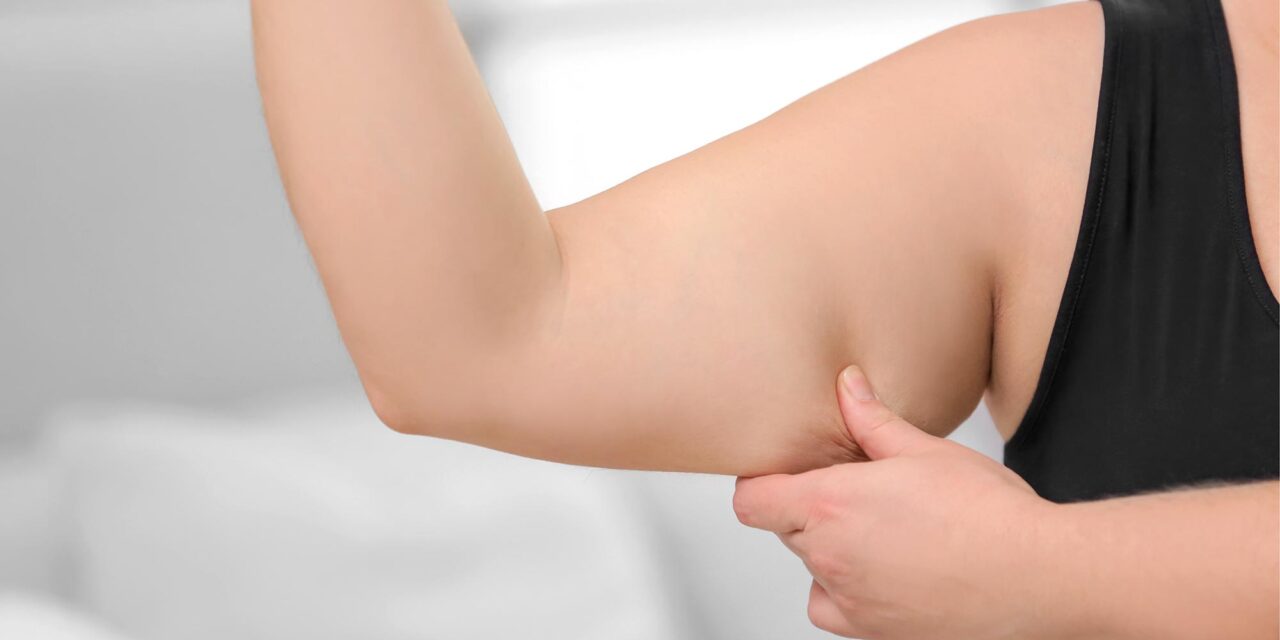
Two groundbreaking studies led by researchers at the Hospital for Special Surgery (HSS) have uncovered critical biological mechanisms that contribute to the development of systemic sclerosis (SSc), commonly known as scleroderma. Published in the March issue of the Journal of Experimental Medicine, the findings offer insights into why this autoimmune disease disproportionately affects women and highlight potential therapeutic targets currently under development.
Scleroderma, a rare and often debilitating condition, leads to fibrosis (tissue hardening) and inflammation, affecting approximately 300,000 people in the United States. About one-third of these cases progress to systemic disease, which can impact vital organs such as the lungs, kidneys, and heart. Notably, women are diagnosed with scleroderma at a rate four times higher than men, though the reasons for this gender disparity had remained largely unknown—until now.
The Role of Genetic Receptors in Scleroderma
One of the studies, led by Dr. Franck Barrat, uncovered that two genetic receptors—Toll-like receptor 7 (TLR7) and Toll-like receptor 8 (TLR8)—are key drivers of plasmacytoid dendritic cells (pDCs), a type of immune cell that fuels chronic fibrosis. These receptors are located on the X chromosome, a factor that plays a crucial role in the disease’s higher prevalence among women.
Typically, one of a female’s two X chromosomes is deactivated in healthy cells. However, the study found that in patients with scleroderma, this process is disrupted. Due to the ability of TLR7 and TLR8 to escape deactivation, these immune cells remain abnormally active, contributing to fibrosis.
“The magnitude of this escape was striking,” said Dr. Barrat. “In healthy individuals, about 10 to 15% of cells evade deactivation, but in scleroderma patients, over 35% of the pDCs exhibited this escape, significantly increasing the expression of these receptors.”
This genetic escape mechanism, the researchers conclude, is a major reason why scleroderma disproportionately affects women.
Why Inflammation Persists in Scleroderma Patients
A second study sought to determine why the body’s natural processes fail to resolve inflammation in scleroderma patients. Normally, after an injury, immune cells infiltrate the affected area and trigger an inflammatory response, which later subsides as healing occurs. However, in scleroderma, this process stalls, leading to prolonged fibrosis.
The researchers identified a cytokine called CXCL4, which is highly expressed in scleroderma patients. Instead of allowing inflammation to subside, CXCL4 prevents immune suppression, keeping pDCs in a persistent state of activation and promoting fibrosis.
“We show that CXCL4 prevents the normal termination of the immune response in the skin,” explained Dr. Barrat. “Essentially, the pDCs are drawn to the fibrosis but remain active due to CXCL4, creating a cycle of chronic inflammation and tissue hardening.”
Potential for New Treatments
Although there is no cure for scleroderma, the studies highlight promising therapeutic strategies. Targeting pDCs could be an effective approach, and several drugs that interfere with these immune cells are already in development.
“This research strongly supports the exploration of pDC-targeting therapies,” said Dr. Barrat. “Some drugs currently in clinical trials for lupus have shown potential in blocking pDCs and preventing skin lesions, and these may prove useful for scleroderma patients as well.”
The studies were a collaborative effort, with contributions from leading researchers and clinicians from the Scleroderma, Vasculitis & Myositis Center of Excellence at HSS, as well as international partners from institutions in France.
Disclaimer:
This article is intended for informational purposes only and does not constitute medical advice. Readers should consult healthcare professionals for diagnosis and treatment of scleroderma or any other medical condition.








{kind=link}