
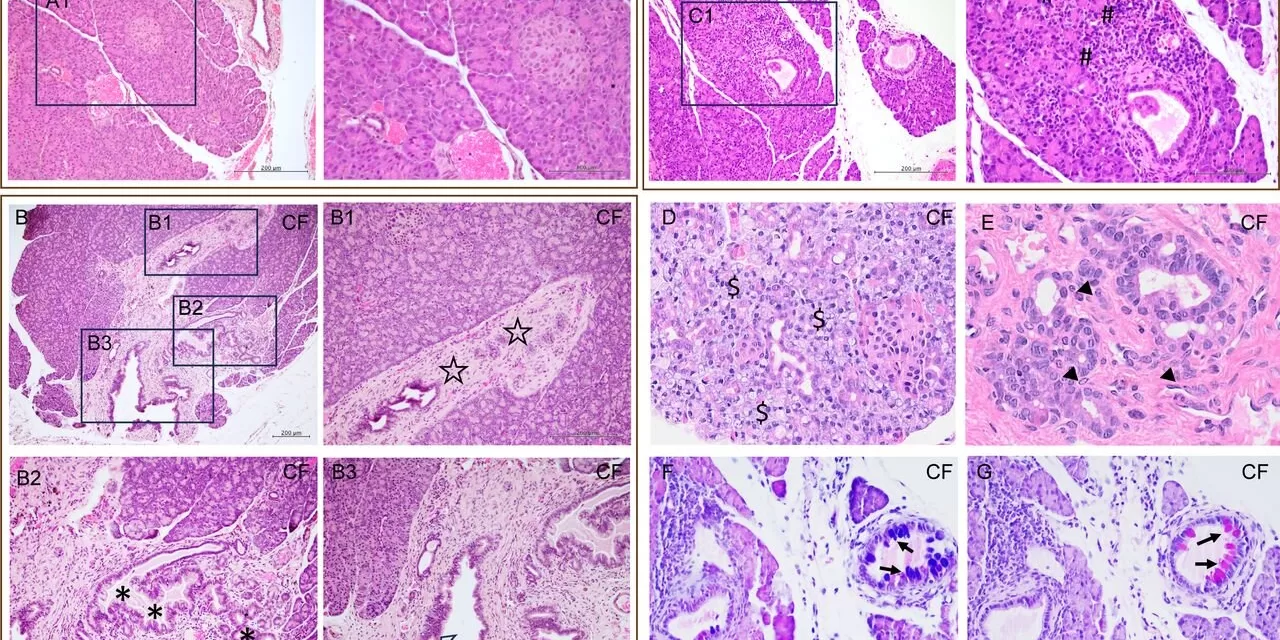
Cystic fibrosis (CF), a life-threatening genetic disorder, affects multiple organs, with pancreatic dysfunction being one of the most critical and often overlooked complications. A groundbreaking study, published in eGastroenterology, introduces young rabbits with CF as a novel and valuable model to investigate CF-related pancreatic endocrine pathology. This new model offers unprecedented insights into cystic fibrosis-related diabetes (CFRD), a condition affecting up to 50% of CF patients.
CF is caused by mutations in the CF transmembrane conductance regulator (CFTR) gene, leading to abnormal chloride and sodium transport across epithelial cells. While advancements like the CFTR modulator drug Trikafta have dramatically improved pulmonary outcomes, pancreatic complications, especially endocrine dysfunction, continue to significantly impact the quality of life for CF patients.
The study, led by Dr. Jie Xu and her team at the University of Michigan Medical School, focuses on the spontaneous development of pancreatic lesions and glucose metabolism abnormalities in CF rabbits, providing new insights into the disease’s underlying mechanisms.
Using CRISPR/Cas9 gene-editing technology, the researchers developed the CF rabbit model, which exhibited hallmark pancreatic changes such as fibrosis, vacuolar degeneration, and mucus-secreting epithelial cell metaplasia. The insulin-producing pancreatic islets in these rabbits were significantly smaller compared to wild-type controls, correlating with lower circulating insulin levels and compromised glucose metabolism.
“Our findings suggest that rabbits with CF replicate key aspects of CF pancreatic disease,” Dr. Xu stated. “This positions them as a valuable model for translational research on CFRD and related conditions.”
One of the study’s notable discoveries was the identification of an indeterminate glucose tolerance (INDET) stage in young CF rabbits, which mirrors early signs of CFRD in human patients. This INDET stage is characterized by delayed glucose clearance and reduced insulin secretion, signaling the onset of diabetes progression. Interestingly, female CF rabbits were found to be more susceptible to INDET-like phenotypes, reflecting the sex-based differences observed in human CFRD prevalence.
This innovative CF rabbit model presents several advantages over existing large-animal models, such as pigs and ferrets, which are often costly and require specialized care. In contrast, rabbits are cost-effective, easier to handle, and commonly used in laboratory settings, making this model more accessible to a broader range of researchers.
“As the CF community turns its attention to age-related and metabolic complications, our rabbit model is becoming increasingly relevant,” Dr. Xu explained. “With improved life expectancy due to CFTR modulators, understanding and managing CFRD will be critical to enhancing the overall quality of life for CF patients.”
The implications of this work extend beyond basic research, as the CF rabbit model could pave the way for the development of targeted interventions. Early-stage therapies addressing the INDET phase may delay or even prevent the onset of full-blown diabetes in CF patients. Additionally, this model provides an opportunity to evaluate emerging treatments aimed at modulating pancreatic inflammation, fibrosis, and insulin production.
Future research will focus on examining the long-term progression of pancreatic disease in CF rabbits and assessing the impact of CFTR modulators like Trikafta on endocrine outcomes.
In conclusion, this study represents a significant advancement in cystic fibrosis research. By harnessing the unique advantages of the CF rabbit model, scientists have the opportunity to deepen their understanding of CF-related pancreatic pathology. This research promises to improve outcomes for the thousands of individuals living with CF and its associated complications.
For more details, refer to the study: Xiubin Liang et al, “Endocrine pathology in young rabbits with cystic fibrosis,” eGastroenterology (2024). DOI: 10.1136/egastro-2024-100102.








{kind=link}