
In a historic breakthrough for sickle cell disease (SCD), the approval and clinical success of novel gene therapies in 2024-2025 have ushered in the first curative treatments for this debilitating genetic blood disorder. These one-time therapies, including lovotibeglogene autotemcel (Lyfgenia) and exagamglogene autotemcel (exa-cel), leverage cutting-edge gene editing to modify patients’ own blood stem cells, dramatically reducing symptoms and resolving vaso-occlusive crises (VOCs) for many patients worldwide. The treatments, now available in the United States and parts of Europe, promise a new standard of care beyond symptom management, offering hope to thousands living with SCD.
Key Findings and Developments
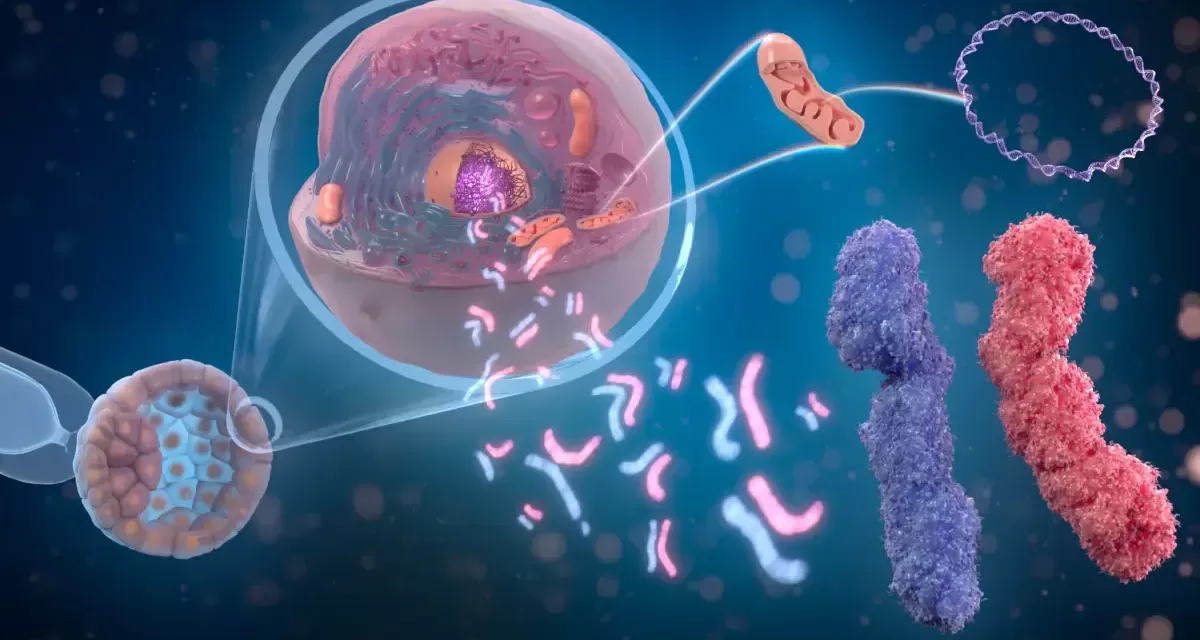
Recent gene therapies approved by the U.S. Food and Drug Administration (FDA) represent a significant milestone for SCD treatment. Exagamglogene autotemcel (exa-cel), approved in December 2023, and lovotibeglogene autotemcel (Lyfgenia), approved shortly thereafter, are autologous cell-based gene therapies that harvest a patient’s own hematopoietic stem cells, edit them to produce healthy hemoglobin, and reinfuse them to the patient in a single dose.
Clinical trial data show remarkable outcomes: 88% of treated patients experienced a complete resolution of vaso-occlusive events between 6 to 18 months post-treatment, which are painful crises caused by sickled red blood cells blocking blood flow. Patients have reported freedom from symptoms for months and even years, transforming disease prognosis.
These therapies work by increasing the production of fetal hemoglobin (HbF), which inhibits the sickling process in red blood cells, improving oxygen transportation and reducing vascular blockages. Unlike previous treatments such as hydroxyurea, painkillers, and blood transfusions that focused on symptom control, gene therapy targets the disease’s underlying genetics for lasting remission.
Expert Commentary
Dr. Anjali Mehta, a hematologist not involved in the clinical trials, explains, “These gene therapies represent a powerful shift from managing sickle cell symptoms to potentially curing the disease by correcting the genetic defect. Using a patient’s own cells also eliminates issues related to donor availability and reduces transplant rejection risks.”
However, she cautions, “While the initial results are promising, long-term safety and accessibility remain challenges. Gene therapy requires sophisticated infrastructure and is currently available to a limited patient population.”
Dr. Raj Patel, a geneticist specializing in blood disorders, adds, “These advances also highlight the importance of continued research into the complex biology of sickle cell disease, including genetic modifiers and environmental factors that influence disease severity.”
Context and Background
Sickle cell disease is a hereditary blood disorder caused by a mutation in the beta-globin gene, leading to the production of abnormal hemoglobin S. This mutation causes red blood cells to deform into a sickle shape, which can obstruct blood flow, causing pain, organ damage, and increased risk of infection. Approximately 100,000 people in the U.S. and millions worldwide, especially in sub-Saharan Africa and India, live with SCD.
Until now, treatment largely focused on symptom relief and prevention of complications, with hydroxyurea being the most common disease-modifying drug. Curative options included hematopoietic stem cell transplantation, which is limited by donor availability and transplant-related risks.
Public Health Implications
The approval of gene therapies like Lyfgenia and exa-cel is a transformative development in public health for communities impacted by SCD. These therapies signal a potential to drastically reduce morbidity and healthcare costs associated with lifelong management and repeated hospitalizations for VOCs.
However, equitable access remains a significant public health concern. These treatments are complex and costly, with accessibility uneven across different regions and socioeconomic groups. Expanding infrastructure, insurance coverage, and support mechanisms will be critical to making these cures broadly available.
Limitations and Counterarguments
Despite their promise, gene therapies for SCD have limitations. The long-term durability of these cures beyond several years is still under study. There are risks associated with the stem cell harvesting and reinfusion process, including potential immune reactions or unforeseen genetic consequences.
Moreover, the high cost and specialized care setting limit current accessibility, especially in low-resource countries where SCD burden is highest. Researchers emphasize complementary approaches such as improved pharmacological agents and ongoing symptom management remain essential until gene therapies become widely available and affordable.
Practical Guidance for Patients and Families
For individuals living with SCD and their families, these advances mean a hopeful future but also the need for informed discussions with healthcare providers. Gene therapy may be a treatment option for eligible patients, typically those aged 12 years and above, with access to specialized centers.
Patients should continue standard care protocols and consult their hematologists about new therapies and clinical trial availability. Preventive care, pain crisis management, and holistic support remain vital components of SCD management today.
Medical Disclaimer
Medical Disclaimer: This article is for informational purposes only and should not be considered medical advice. Always consult with qualified healthcare professionals before making any health-related decisions or changes to your treatment plan. The information presented here is based on current research and expert opinions, which may evolve as new evidence emerges.







{kind=link}