
A groundbreaking study has taken a significant step toward understanding Rett syndrome, a rare genetic disorder affecting approximately 1 in 10,000 newborn girls. This devastating condition is characterized by a sudden regression in development around one year of age, leading to the loss of acquired language and motor skills and resulting in severe cognitive impairment. Rett syndrome is primarily caused by mutations in the MeCP2 gene, a critical regulator of neuronal development in the brain.
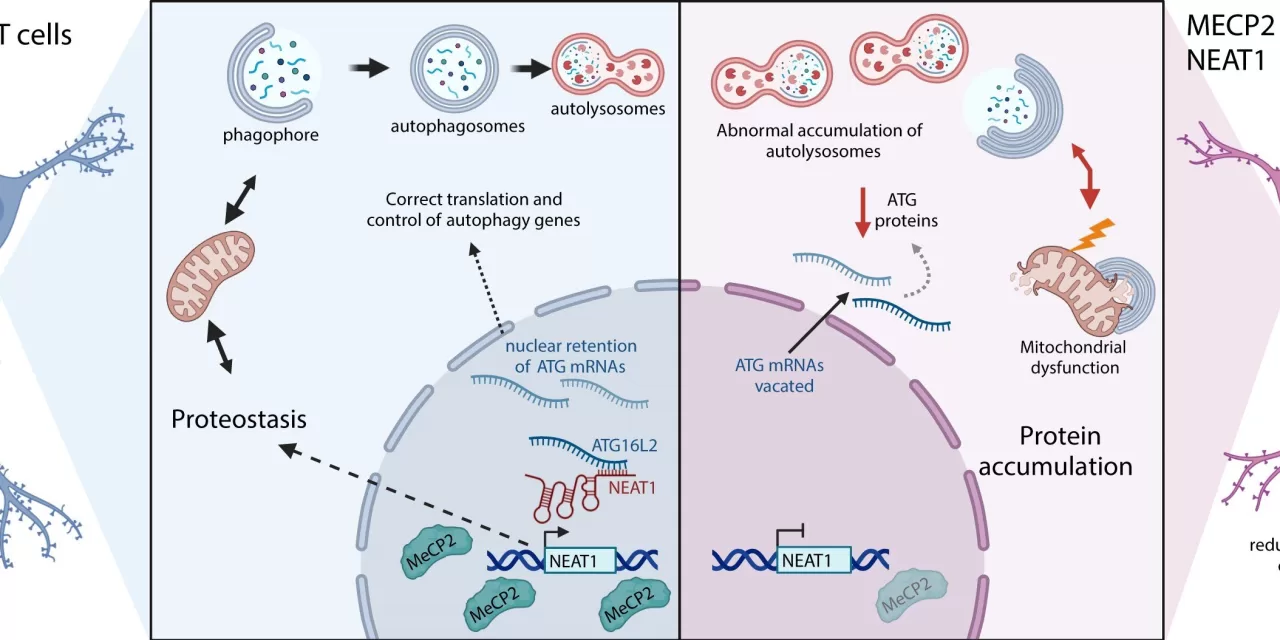
Due to the gene’s crucial role as a master controller, deciphering the exact mechanisms by which MeCP2 mutations cause the numerous brain cell abnormalities—such as structural changes and synaptic imbalances—has been a formidable challenge for researchers. However, a new study conducted by the Regulatory RNA and Chromatin Lab at the Josep Carreras Institute sheds light on one of these key alterations: autophagy malfunction.
The Role of Autophagy in Cellular Maintenance
Autophagy, derived from the Greek term meaning “self-eating,” is a vital cellular process that helps eliminate obsolete components, such as unnecessary proteins and organelles that are no longer required. This self-maintenance system ensures that cells remain healthy and function properly. However, defects in autophagy can lead to the accumulation of protein aggregates and other intracellular deficiencies, which contribute to abnormal cellular functions and serious consequences in neurological disorders like Rett syndrome.
Key Findings: The Link Between MeCP2 and NEAT1
The research, first-authored by Dr. Edilene Siqueira and recently published in Nucleic Acids Research, employed cutting-edge genetic technologies, including single-cell RNA sequencing and CRISPR/Cas9 gene editing. The study found that mutations in the MeCP2 gene led to the depletion of a long non-coding RNA molecule known as NEAT1.
NEAT1 plays a crucial role in regulating the autophagy process by forming direct RNA-RNA interactions with key components of the autophagy machinery and determining their cellular localization. Consequently, NEAT1 deficiency contributes to the cellular dysfunctions observed in Rett syndrome.
Potential for Therapeutic Advances
In a promising breakthrough, the researchers demonstrated that restoring NEAT1 levels in in vitro models of Rett syndrome could reverse some of the cellular abnormalities associated with the disease. This finding opens the door to exploring new therapeutic strategies targeting autophagy regulation in Rett syndrome patients.
The study was a collaborative effort among the Regulatory RNA and Chromatin Group led by Dr. Sònia Guil, the Cancer Epigenetics Laboratory headed by Dr. Manel Esteller at the Josep Carreras Institute, as well as researchers from the Bellvitge Biomedical Research Institute (IDIBELL) and the Hospital Sant Joan de Déu.
Future Implications
This research represents an important step forward in unraveling the molecular mechanisms behind Rett syndrome and highlights the potential of RNA-based therapies to address neurodevelopmental disorders. While further studies are needed to translate these findings into clinical treatments, this discovery paves the way for novel interventions that could one day improve the lives of individuals affected by this debilitating condition.
Reference: Edilene Siqueira et al., NEAT1-mediated regulation of proteostasis and mRNA localization impacts autophagy dysregulation in Rett syndrome, Nucleic Acids Research (2025). DOI: 10.1093/nar/gkaf074
Disclaimer: This article is for informational purposes only and does not constitute medical advice. While the findings are promising, further research and clinical trials are necessary before any potential treatments become available. Individuals seeking medical guidance on Rett syndrome should consult healthcare professionals.









{kind=link}